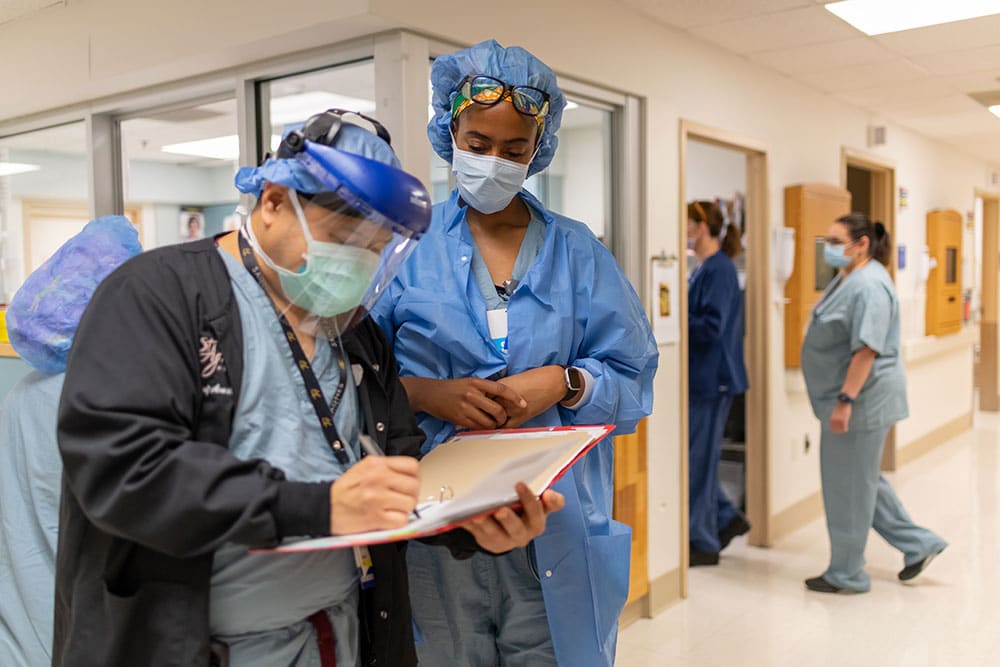
Healthy Hospitals.
Healthy Communities.
The Maryland Hospital Association serves hospitals and health systems through collective action to shape policies, practices, and performance, advancing health care and the health of all Marylanders.

Caring for Communities
To MHA hospitals and health systems, caring for their community extends far beyond the hospital walls. Each member hospital and health system takes a holistic approach in neighborhoods and with local partners to address the needs of their community.
In one year, Maryland hospitals care for



Healthy Hospitals are the Foundation of Healthy Communities
MHA’s member-driven priorities in 2025 are:
-
Healthy Hospitals. Healthy Communities.
Support policies that strengthen hospitals’ ability to deliver high-quality care, ensuring healthier communities by expanding access to care and promoting financial sustainability. -
Ensure Access to Care
Establish a systemic response to pediatric overstays by building bed capacity across the state, creating pathways of accountability and transparency to support youth and families in crisis, and improving processes that hinder timely access to care -
Protect and Improve Maryland’s Medical Liability Climate
Until comprehensive reform can be achieved, protect against further erosion in the state’s existing liability climate so Maryland remains a premiere location for medical practice. -
Strengthen Payer Accountability
Create a system for reviewing payer denials that refines data disclosures and ensures data integrity, enhances payer denial transparency, and reduces denial rates, while examining factors that contribute to excessive denial rates such as the use of AI in claims reviews and prior- authorization requirements. -
Secure the Future of Telehealth Programs
Permanently preserve telehealth flexibilities successfully expanded during the pandemic to ensure patients and providers have continued access to a crucial means of care delivery.

Building a Healthy Workforce
Care starts with the professionals who serve patients and their communities both in and outside the hospital. JoinMdHealth, an initaitve launched by MHA and our members, connects current and future hospital employees with education and career opportunities that match their strengths and goals.